Alpha-mannosidosis: An ultra-rare disease
that affects all ages
Alpha-mannosidosis is a very rare condition known as a lysosomal storage disorder. It is a chronic disease inherited through genes passed down from parents to their children.

Alpha-mannosidosis affects about
1 in 500,000 to 1 in 1,000,000 live births.
Alpha-mannosidosis at a glance:
- Alpha-mannosidosis is what’s considered a “progressive disease,” meaning it changes or gets worse over time. Children may have more issues with hearing and mental function, while people in their 20s and 30s may have more problems with bones, joints, and muscles
- These effects range from mild to severe. Severe disease will be more noticeable at birth, can develop rapidly, and unfortunately is life-threatening. Mild to moderate cases progress more slowly over time
- If untreated, alpha-mannosidosis will likely continue to progress in severity––which is why it’s critical to start treatment early
Who does this disease usually affect?
Usually diagnosed in infants or teens, alpha-mannosidosis may first be recognized due to developmental delays or when a young person starts to show symptoms. It is then confirmed through enzymatic and/or genetic testing.
What happens in the body?
 |
 |
 |
||||||
|
An unusual gene mutation is inherited, causing alpha-mannosidosis |
|
|
If untreated, alpha-mannosidosis will likely continue to progress as excess oligosaccharides build up in the cells of the body. That’s why it’s critical to start treatment early. This is especially important with an ultra-rare disease like alpha-mannosidosis because treatment may have already been delayed due to the difficulty of diagnosis.
Early diagnosis of alpha-mannosidosis is key to help slow its progression.
What may happen over time?
The long-term effects of alpha-mannosidosis can make movement more challenging and could lead to disabilities requiring the assistance of a caregiver, especially as an adult.
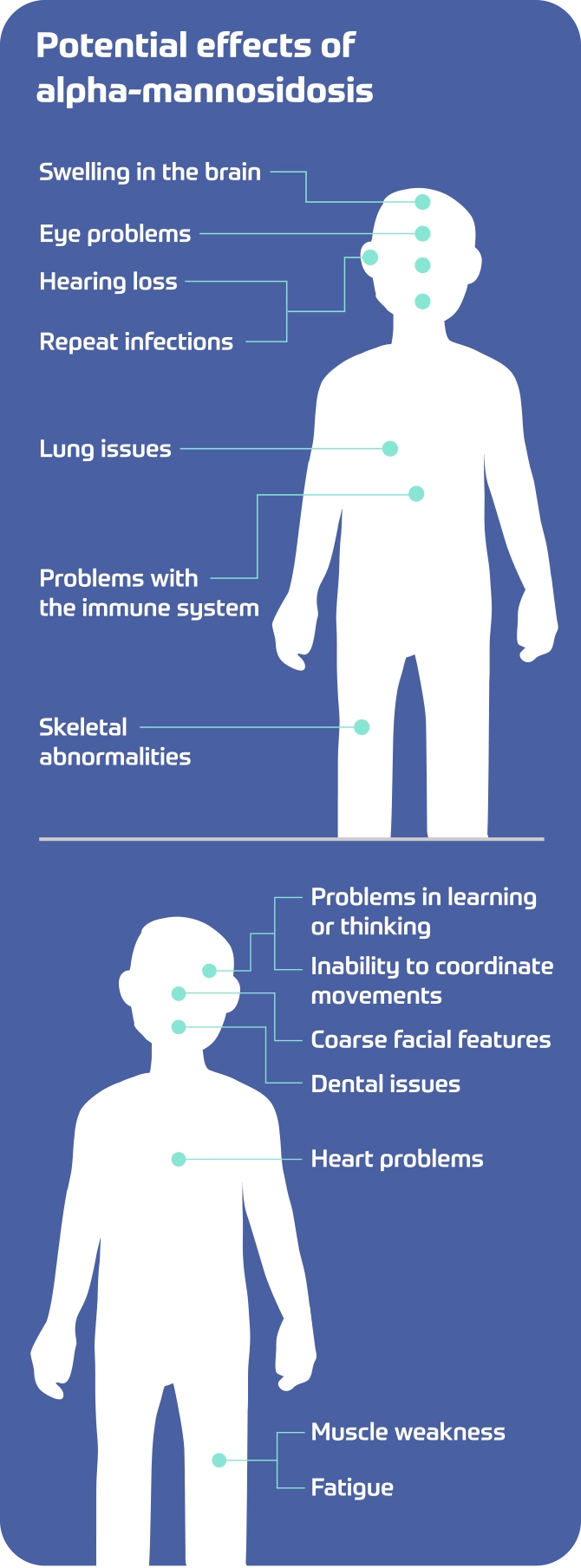
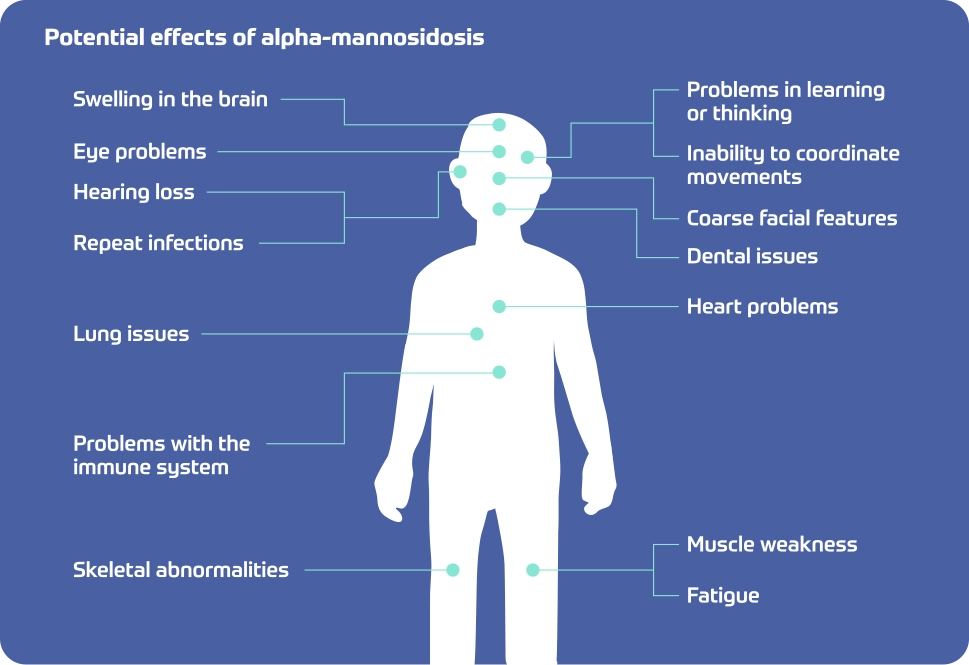
Potential effects of alpha-mannosidosis
- Swelling in the brain
- Eye problems
- Hearing loss
- Repeat infections
- Lung issues
- Problems with immune system
- Skeletal abnormalities
- Problems in learning and thinking
- Inability to coordinate movements
- Coarse facial features
- Dental issues
- Heart problems
- Muscle weakness
- Fatigue
A progressive disease is one that gets worse over time.
You or your loved one may not be at the point where these changes are really noticeable. But progression could happen if you wait too long to get help.
What may people experience?
There are 3 types of alpha-mannosidosis:
Mild
Symptoms: Muscle weakness, mild to moderate mental disability
Progression: Slow. Progression may stop beyond teen years
Moderate
Symptoms: Impaired ability to coordinate movements (ataxia) by the age of 20-30
Progression: Slow to moderate
Severe
Symptoms: Problems in learning or thinking, brain swelling, ataxia, swelling of the liver and spleen, skeletal problems, and coarse facial features
Progression: Rapid
The severe type will be more noticeable at birth, develops rapidly, and is unfortunately life-threatening. The mild and moderate types are slower to progress over time.
What treatments are available for alpha-mannosidosis?
Until now, there hasn't been an FDA-approved treatment option specifically designed for alpha-mannosidosis. Symptom control has been the most common way to manage the effects.* This can include:

Antibiotics to suppress infections

Physical therapy for muscle weakness

Hearing aids and tubes to improve hearing

Tubes (shunts) to drain excess fluid within the tissue that surrounds the brain and spinal cord
*Bone marrow transplants have been used to treat alpha-mannosidosis. However, they are not always an option because some patients are diagnosed too late and others cannot find appropriate donors.
Although there is no cure, you'll be happy to learn that,
for the first time ever, there is an enzyme replacement therapy available.
Introducing Lamzede.